


从头算分子动力学揭秘咪唑类缓蚀剂在铜/水界面的吸附机制
铜因优异的导电性、导热性和延展性,被广泛应用于多个工业领域,但在水和空气环境中易发生腐蚀,造成经济损失与安全风险。咪唑类有机缓蚀剂因高效、低成本成为铜腐蚀防护的常用选择,但其在真实水相环境中的吸附机制(尤其是水分子的影响)尚未被精准揭示,制约了高性能缓蚀剂的理性设计。
北京科技大学张达威团队联合中科院宁波材料所等单位在Corrosion Science发表题为Understanding the adsorption of imidazole corrosion inhibitor at the copper/water interface by ab initio molecular dynamics的研究成果。该团队采用从头算分子动力学(AIMD)模拟结合密度泛函理论(DFT)计算,首次在原子尺度上阐明了咪唑分子在铜/水界面的吸附行为及水分子的调控作用,为缓蚀剂分子设计提供了关键理论依据。
研究核心:AIMD揭示铜/水界面的吸附本质
研究团队以咪唑分子为缓蚀剂模型,铜(111)表面为研究对象,构建铜/水界面模型,通过AIMD模拟捕捉动态吸附过程,核心聚焦“咪唑-铜表面-水分子”三者的相互作用,破解传统真空模拟与真实环境脱节的难题。
1. 模拟体系与计算方法
模型构建:采用4×4周期性超胞的Cu (111) 表面(6个原子层),构建铜/水界面体系(水密度1 g/cm³),分别设置真空环境与水相环境对照组,咪唑分子初始为水平和垂直两种吸附构型;
计算方法:
结构优化与电子结构计算:基于DFT的CP2K和VASP程序,采用PBE泛函与Grimme D3色散校正,精准描述电子转移与化学键作用;
动力学模拟:采用第二代Car-Parrinello分子动力学(SGCPMD),模拟温度330 K(精准描述水的氢键网络),模拟时长25 ps(5 ps平衡+20 ps数据采集);
分析手段:通过吸附能、差分电荷密度Bader电荷分析、径向分布函数(RDF)等,量化吸附强度与微观作用。
2.吸附构型与稳定性:垂直吸附为优势构型
真空与水相环境中,咪唑分子均优先采用垂直吸附构型:通过咪唑环上不饱和N3原子与Cu表面顶位原子结合,N3-Cu键长约2.07 Å,吸附能为-1.51 eV(真空)和-1.25 eV(水相);
水平吸附构型在水相中不稳定,会自发转变为垂直构型,原因是水平构型中咪唑环与水分子的疏水作用及空间位阻不利于吸附稳定;
水相中的吸附能略高于真空环境(更正值),这是由于咪唑分子与水分子形成氢键,削弱了其与铜表面的相互作用。
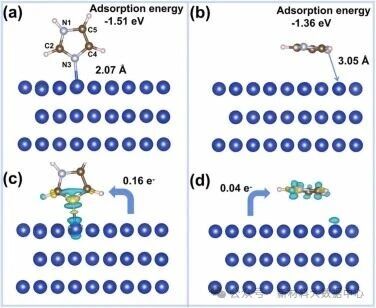
图1 咪唑分子在Cu(111) 表面的吸附结构: (a)垂直吸附构型; (b)水平吸附构型; 咪唑分子在Cu(111) 表面吸附的电荷密度差图: (c)垂直吸附构型; (d)水平吸附构型
3. 水分子的三重调控作用
这是研究的核心突破,首次明确水分子并非单纯溶剂,而是通过多重机制影响吸附过程:
竞争吸附与界面水密度调控:咪唑优先吸附于铜表面,排挤第一层化学吸附水,使第一层水分子数从2.14降至0.97,表面水覆盖率从0.61 ML 降至0.42 ML,减少腐蚀介质与铜表面的接触;
氢键网络破坏:咪唑分子的疏水基团(C-H 键)产生空间位阻,破坏界面水分子的连续氢键网络,减缓腐蚀性H⁺离子的迁移速率,抑制腐蚀反应;
水停留时间缩短:吸附咪唑后,界面化学吸附水与铜表面的平均停留时间从800 fs骤降至97 fs,降低了水分子参与腐蚀反应的概率。
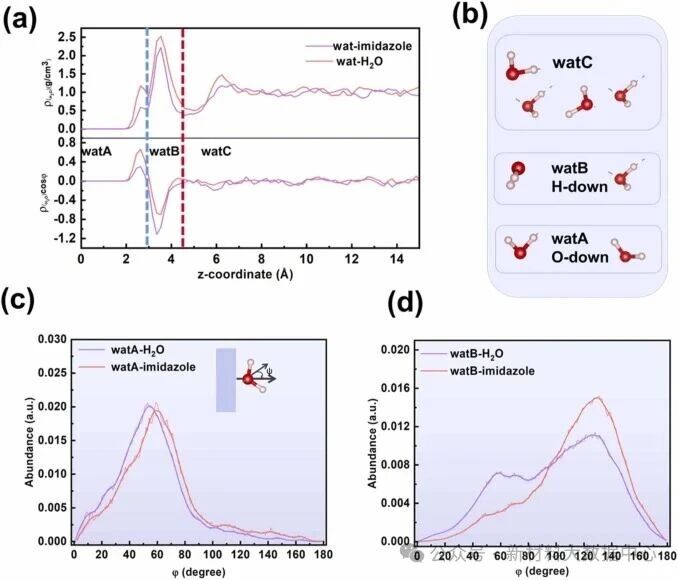
图2 (a)有无咪唑条件下铜/水界面的界面水密度及偶极取向分布对比; (b)水分子取向分布图; (c)水分子A(watA)的归一化偶极角分布; (d)水分子 B(watB)的归一化偶极角分布
4. 电子转移与吸附本质
差分电荷密度分析显示,垂直吸附时电子从咪唑分子向铜表面转移(转移量0.16 e⁻),N3 原子与相邻Cu原子形成强相互作用,电子云重叠明显;
水平吸附时电子转移量仅0.04 e⁻,相互作用较弱,进一步验证垂直构型的优势;
咪唑环上N1原子的H原子与水分子的O原子形成氢键(键长1.7 Å),这是水相吸附能降低的关键原因。
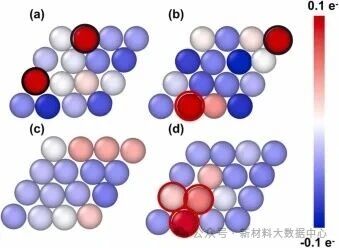
图3 铜表面原子电荷分布: (a)未添加咪唑; (b)添加咪唑; 铜表面原子的平均电荷分布: (c)未添加咪唑; (d)添加咪唑
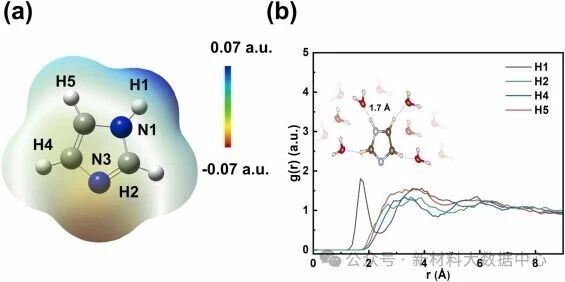
图4 (a)咪唑的静电势分布; (b)咪唑中氢原子与水分子中氧原子的径向分布函数(RDF)术语补充说明
关键发现:缓蚀机制的原子尺度解析
1. 吸附优先级:咪唑>水分子
咪唑与铜表面的吸附亲和力强于水分子,能在竞争吸附中占据优势位点,形成致密的分子膜,物理阻挡腐蚀介质入侵,这是其发挥缓蚀作用的核心前提。水相环境中,咪唑虽取向有小幅摆动,但仍保持垂直吸附构型,N3-Cu键长与真空环境接近,吸附稳定性良好(图5)。
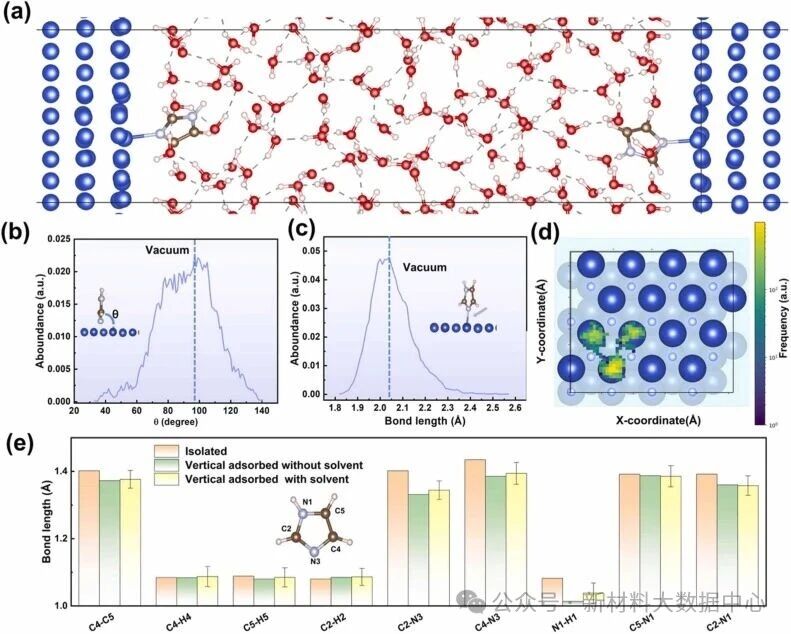
图5 (a)显性水环境下的分子吸附结构(红色球代表氧原子); (b)咪唑与铜表面的取向角; (c)咪唑中N3原子与相邻铜原子的键长; (d)咪唑中氮原子在xy平面的投影; (e)不同条件下咪唑分子内的键长
2. 缓蚀协同效应
咪唑的缓蚀效果源于双重机制:
物理屏障:分子吸附层阻挡O₂、H₂O等腐蚀介质与铜表面接触;
微观调控:通过破坏氢键网络、缩短水停留时间,抑制腐蚀性离子迁移与电化学反应动力学,从根源上减缓腐蚀进程。
3. 结构-性能关联
咪唑分子的缓蚀效率与其结构密切相关:
含孤对电子的N原子是吸附锚定位点,决定了与铜表面的结合强度;
分子的疏水基团(如咪唑环上的C-H)通过空间位阻调控界面水环境,影响吸附稳定性与缓蚀效果(图4)。
研究价值:为缓蚀剂设计提供理论范式
方法创新:首次采用AIMD模拟精准描述铜/水界面的动态吸附过程,突破传统真空模拟的局限性,实现对真实环境下吸附机制的原子尺度揭秘;
机制突破:明确水分子在吸附过程中的 “双重作用”——既通过氢键削弱咪唑与铜的结合,又通过被排挤和氢键网络破坏间接增强缓蚀效果,修正了“水仅为溶剂”的传统认知;
设计指导:提出缓蚀剂分子的理性设计准则——需具备强吸附位点(如含孤对电子的N、O原子)与适度疏水基团,平衡与金属表面的结合强度和对界面水环境的调控能力;
跨领域启示:为其他唑类(苯并三唑、噻二唑)缓蚀剂的机制研究提供通用方法,也为金属 / 溶液界面的吸附行为研究提供参考。
该研究通过从头算分子动力学与密度泛函理论的结合,首次在原子尺度上阐明了咪唑缓蚀剂在铜/水界面的吸附构型、电子转移规律及水分子的调控机制,深刻揭示了缓蚀作用的本质。研究成果不仅解决了传统模拟与真实环境脱节的关键问题,更为高性能缓蚀剂的分子设计提供了精准的理论指导,有望推动腐蚀防护技术从“经验筛选”向“理性设计”跨越。
未来研究可进一步拓展至咪唑衍生物(如取代基修饰),探究结构改性对吸附性能的影响,同时结合实验表征(如表面增强拉曼光谱、电化学测试)验证模拟结论,实现理论与实验的深度融合。
论文原文:
Understanding the adsorption of imidazole corrosion inhibitor at the copper/water interface by ab initio molecular dynamics - ScienceDirectX. Guo, X. Zhang, L. Ma, Y. Li, J. Le, Z. Fu, L. Lu, D. Zhang. Understanding the adsorption of imidazole corrosion inhibitor at the copper/water interface by ab initio molecular dynamics[J]. Corrosion Science, 2024, 236: 112237.
原文地址:
https://www.sciencedirect.com/science/article/pii/S0010938X24004323?via%3Dihub